[Most Recent Entries] [Calendar View] [Friends]
Below are the 4 most recent journal entries recorded in
wasserstrahl's LiveJournal:
| Sunday, November 15th, 2015 | |
| 11:07 pm | CP2K: erratum Пара уточнений по предыдущим постам. 1. Во-первых, более тщательный анализ показал, что центрирование молекулы в ячейке необходимо только в частном случае вэйвлетного решателя уравнения Пуассона, в общем случае это необязательно, плотность на границе ячейки может быть ненулевой (получаются псевдо-периодические краевые условия --- короче, оно работает и без соотв. инструкции, давая на выходе ту же электронную энергию, что и после центрирования). Убирать из примера не стал, но добавил уточнение в пост. 2. Провтыкал, что HTML воспринял инструкцию ¢ как управляющую даже несмотря на тэг PRE, исправил. 3. Оказалось, что я закрыл в жжшном посте с разбором полетов комментирование. Это случайность, я забыл его открыть после очередного исправления, это связано с нюансами экспорта с тифаретника на жж. Уже открыто. И да, есть ли у вас пожелания по выбору веселых молекул для следующих серий? VX? метамфетамин? TNT?) |
| 12:30 am | CP2K: анализ результатов оптимизации LSD В продолжение к http://lj.rossia.org/users/wasserst 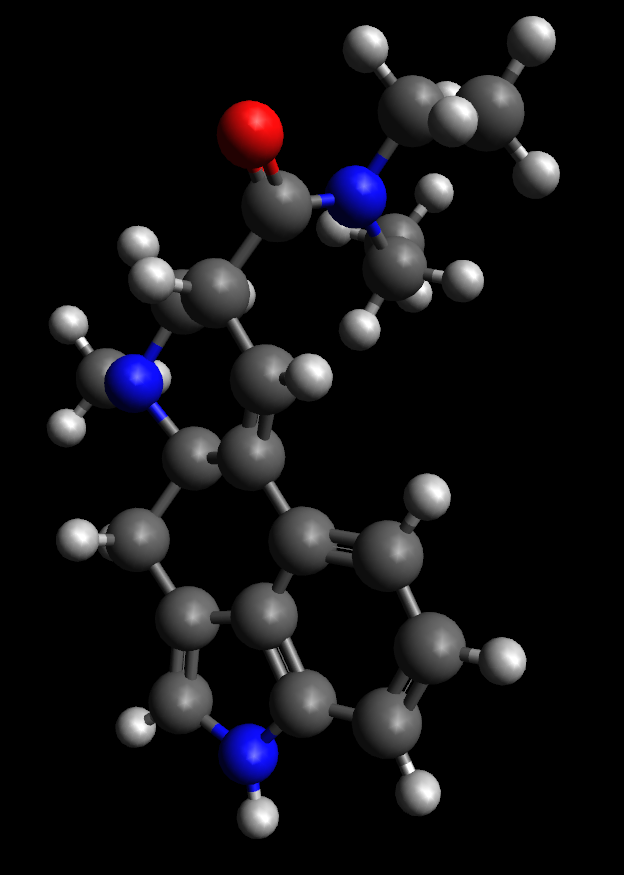 Итак, что мы получили на выходе (файлы из моего расчета приведены по ссылке https://mega.nz/#F!oQJ00bjY!wAFcNt6f5eC 1. "Протокол" расчета (output-файл) opt_LSD.out, содержащий информацию о ходе и результатах расчета. 2. Файл opt_LSD-pos-1.xyz, содержащий траекторию изменения геометрии молекулы в процессе оптимизации. 3. Несколько файлов *.restart --- это файлы "сохранения", позволяющие в случае прерывания выполнения программы перезапустить расчет не с начала, а с одного из последних достигнутых этапов. 4. Соответствующие им файлы *.wfn --- это волновые функции, полученные в результате решения ССП, которые также могут быть прочитаны, проанализированы и использованы. 5. Файл opt_LSD-BFGS.Hessian, содержащий приближение гессиана, полученное в ходе оптимизации геометрии с помощью алгоритма Бройдена-Флетчера-Гольдфарба-Шанно (BFGS), используемого в CP2K по умолчанию. Output-файл.Начинается преамбулой с указанием ряда технической информации, включая версию программы, флаги компиляции, некоторые параметры расчета, включая исходную геометрию, выбранный метод и т.д. Далее начинается процедура оптимизации геометрии: ******************************************************************************* *** STARTING GEOMETRY OPTIMIZATION *** *** BFGS *** ******************************************************************************* Последующая часть output'а состоит из набора однообразных фрагментов, каждый из которых соответствует очередному шагу поиска равновесной геометрии. Техническая информация:
--------------------------
OPTIMIZATION STEP: 2
--------------------------
DISTRIBUTION OF THE NEIGHBOR LISTS
Total number of particle pairs: 1194
Total number of matrix elements: 7413
Average number of particle pairs: 1194
Maximum number of particle pairs: 1194
Average number of matrix element: 7413
Maximum number of matrix elements: 7413
Number of electrons: 126
Number of occupied orbitals: 63
Number of molecular orbitals: 63
Number of orbital functions: 121
Number of independent orbital functions: 121
Parameters for the always stable predictor-corrector (ASPC) method:
ASPC order: 0
B(1) = 2.000000
B(2) = -1.000000
Extrapolation method: ASPC
Итеративное решение ССП и его результаты:
SCF WAVEFUNCTION OPTIMIZATION
----------------------------------- OT ---------------------------------------
Minimizer : CG : conjugate gradient
Preconditioner : FULL_SINGLE_INVERSE : inversion of
H + eS - 2*(Sc)(c^T*H*c+const)(Sc)^T
Precond_solver : DEFAULT
Line search : 2PNT : 2 energies, one gradient
stepsize : 0.08000000 energy_gap : 0.08000000
eps_taylor : 0.10000E-15 max_taylor : 4
----------------------------------- OT ---------------------------------------
Step Update method Time Convergence Total energy Change
------------------------------------------------------------------------------
1 OT CG 0.80E-01 0.6 0.00499787 -132.9638373426 -1.33E+02
2 OT LS 0.25E+00 0.0 -132.9766523882
3 OT CG 0.25E+00 0.2 0.00248243 -132.9877867426 -2.39E-02
4 OT LS 0.27E+00 0.2 -132.9941400844
5 OT CG 0.27E+00 0.2 0.00111336 -132.9941768872 -6.39E-03
6 OT LS 0.25E+00 0.7 -132.9953375248
7 OT CG 0.25E+00 0.2 0.00057071 -132.9953490620 -1.17E-03
8 OT LS 0.26E+00 0.1 -132.9956695813
9 OT CG 0.26E+00 0.2 0.00029537 -132.9956701023 -3.21E-04
10 OT LS 0.22E+00 0.2 -132.9957389738
11 OT CG 0.22E+00 0.2 0.00013662 -132.9957418191 -7.17E-05
12 OT LS 0.23E+00 0.2 -132.9957580814
13 OT CG 0.23E+00 0.2 0.00006623 -132.9957581399 -1.63E-05
14 OT LS 0.24E+00 0.1 -132.9957620855
15 OT CG 0.24E+00 0.2 0.00003683 -132.9957620888 -3.95E-06
16 OT LS 0.31E+00 0.2 -132.9957635908
17 OT CG 0.31E+00 0.2 0.00002360 -132.9957636744 -1.59E-06
18 OT LS 0.31E+00 0.1 -132.9957643309
19 OT CG 0.31E+00 0.2 0.00001652 -132.9957643309 -6.57E-07
20 OT LS 0.28E+00 0.2 -132.9957646181
21 OT CG 0.28E+00 0.2 0.00001146 -132.9957646215 -2.91E-07
22 OT LS 0.29E+00 0.2 -132.9957647646
23 OT CG 0.29E+00 0.3 0.00000792 -132.9957647647 -1.43E-07
*** SCF run converged in 23 steps ***
Core-core repulsion energy [eV]: 27310.25943600658138
Core Hamiltonian energy [eV]: -5778.36888981168067
Two-electron integral energy [eV]: -50301.77870500837162
Electronic energy [eV]: -30929.25824231586739
Total energy [eV]: -3618.99880630928556
Atomic reference energy [eV]: 3619.16398738078260
Heat of formation [kcal/mol]: 3.80916630197567
outer SCF iter = 1 RMS gradient = 0.79E-05 energy = -132.9957647647
outer SCF loop converged in 1 iterations or 23 steps
Далее следует анализ распределения зарядов по Малликену (сами посмотрите), после него --- суммарная информация по шагу и сравнение результатов с допусками. Этап расчета градиента при таких настройках вывода не озвучивается. ENERGY| Total FORCE_EVAL ( QS ) energy (a.u.): -132.995764830868666 -------- Informations at step = 2 ------------ Optimization Method = BFGS Total Energy = -132.9957648309 Real energy change = -0.0030547509 Predicted change in energy = -0.0061894953 Scaling factor = 0.0000000000 Step size = 0.1108009430 Trust radius = 0.4724315332 Decrease in energy = YES Used time = 5.079 Convergence check : Max. step size = 0.1108009430 Conv. limit for step size = 0.0030000000 Convergence in step size = NO RMS step size = 0.0230440603 Conv. limit for RMS step = 0.0015000000 Convergence in RMS step = NO Max. gradient = 0.0102268176 Conv. limit for gradients = 0.0004500000 Conv. for gradients = NO RMS gradient = 0.0033158036 Conv. limit for RMS grad. = 0.0003000000 Conv. for gradients = NO --------------------------------------------------- Поскольку критерии сходимости не достигнуты, начинается следующий шаг. После достижения критериев выполняется еще один процесс решения ССП в равновесной точке, выводит финальную электронную энергию: ENERGY| Total FORCE_EVAL ( QS ) energy (a.u.): -132.999805724373459 После чего выводит статистическую информацию, подборку ссылок на статьи по методам, использованным в расчете (важно и удобно!) и завершается. Траектория изменения геометрии.Файл opt_LSD-pos-1.xyz состоит из набора XYZ-файлов (слепленных в один), каждый из которых описывает геометрию на соответсвующем шаге оптимизации. Этот файл можно просмотреть в текстовом редакторе (и, например, скопировать из него последнюю --- равновесную --- геометрию), а также можно открыть в Avogadro и анимировать движение к равновесию: для этого нужно выполнить avogadro opt_LSD-pos-1.xyz, после чего выбрать Меню --- Расширения --- Анимация и в появившемся окошке открыть тот же файл еще раз. После чего можно автоматически (работает не везде) или вручную ползунком анимировать изменение геометрии в процессе оптимизации. Можно также сохранить это как видео, только нужно установить на систему пакеты povray и mencoder. Пример видео приведен в каталоге с файлами расчета. |
| Saturday, November 14th, 2015 | |
| 3:15 pm | CP2K: первый пример. Полуэмпирическая оптимизация молекулы LSD Итак, начнем разбирать примеры с простого и приятного -- полуэмпирики. Почему именно с архаичного и забытого многими метода? Прежде всего потому, что для запуска рабочего примера нам понадобится настроить минимальное количество опций, т.к. многие из них определяются методом. Соответственно, меньше теории перед тем, как запустить реальное вычисление и посмотреть на его работу. Все файлы я выкладываю на облачное хранилище MEGA, они будут доступны по ссылкам в постах. Если удобнее куда-то в другое место --- напишите. В частности, файлы для данного расчета лежат здесь: https://mega.nz/#F!oQJ00bjY!wAFcNt6f5eC Для работы CP2K в минимальном варианте нужен только один файл, содержащий управляющие опции для работы программы. Этот файл также должен содержать исходную геометрию исследуемой системы --- координаты атомов. В принципе, координаты атомов могут быть размещены в дополнительном внешнем файле, но не будем пока что усложнять конструкцию. Конфигурационный файл CP2K состоит из секций (вкладываемых друг в друга) и параметров. Начало секции объявляется амперсандром & с названием секции, завершение -- амперсандом со словом end и необязательным названием секции, например: &globalИли: &globalНесмотря на то, что название секции после &end необязательно, его лучше указывать, чтобы не запутаться. Параметры указываются как имя и значение (для некоторых параметров необязательное), разделенные пробелами. &globalВосклицательный знак используется для объявления комментариев (все содержимое от восклицательного знака до конца строки игнорируется). Регистр букв не имеет значения --- по традиции почему-то обычно пишут прописными, но я лично предпочитаю строчные. Набор возможных настроек обширный, но настраивать, разумеется, нужно не все. Большинство параметров имеют относительно разумные значения по умолчанию. Таким образом, мы изменяем параметры там, где: --- Мы непосредственно хотим поменять поведение программы; --- Где значения по умолчанию нас не устраивают; --- Где значения по умолчанию не совместимы с другими настройками расчета, которые нам нужны (к сожалению, CP2K не умеет менять одни параметры в зависимости от значений других). Обычно для учебных расчетов пользователи CP2K почему-то предпочитают кластеры воды, но оставим это уныние и попробуем оптимизировать что-то повеселее --- например, молекулу LSD-25. Для получения начальных координат молекулы воспользуемся свободной программой-редактором Avogadro, которую можно скачать с официального сайта или установить из репозитория. В Avogadro нужно нарисовать молекулу по структурной формуле, после чего оптимизировать ее геометрию молекулярной механикой (Меню -- Расширения -- Оптимизировать геометрические параметры, повторять пока молекула не перестанет изменяться), результат сохранить в файл в формате XYZ (*.xyz) (в каталоге с файлами расчета результат сохранен как LSD_mm.xyz). Ниже привожу комментированный файл конфигурации (input-файл). Звездочки означают пояснения после текста файла. ( Конфиг и пояснения ) |
| 12:13 am | CP2K: квантовая химия "для кающихся грешников"(с) Разгребаюсь потихоньку с CP2K. Совершенно примечательная программа для квантовохимических расчетов. Свободная и бесплатная, от перечня возможностей начинается бурное слюноотделение, но уровень документирования... просто катастрофически ужасен. Есть reference manual с кратким описанием параметров, есть набор собственных тестов, несколько туториалов и упражнений по отдельным вопросам, плюс гугл-группа (весьма живая). Влазить в этот трэш без предварительного опыта с какой-нибудь более вменяемой программой настоятельно не рекомендуется. Ну а я попробую изобразить небольшую серию постов с примерами и объяснениями, по мотивам и итогам, может кому поможет. Итак, для начала о том, как поставить это себе на комп. В Debian и особо мною любимом LMDE делается, как и полагается, линейно через apt-get install cp2k (cp2k есть в стандартном репозитории Debian). Однако, если нужна более свежая версия (в том числе экспериментальная), а также если хочется использовать CUDA (да, cp2k это умеет), то нужно ставиться из исходников. В системе нужно поставить следующие пакеты: subversion gfortran gcc make libopenblas-dev libfftw3-dev libint-dev libxc-dev g++ (названия указаны для пакетной базы Debian, все присутствуют в репозитории. UPD 16.01.2016: добавил g++). Для CUDA нужен также nvidia-cuda-toolkit. На самом деле часть этих пакетов опциональна, кроме того, можно использовать еще некоторые дополнительные библиотеки, которых нет в репозитории --- я перечислил все, что можно выжать из обычного репозитория Debian. Отдельное замечание о libopenblas-dev: при установке этот пакет через механизм update-alternatives изменяет конфигурацию системы таким образом, чтобы функции динамически подключаемой библиотеки BLAS на системе выполняла именно OpenBLAS. Если вы устанавливали OpenBLAS ранее или пользуетесь другими библиотеками --- Инструкции по получению исходников приведены на оф.сайте: http://www.cp2k.org/download Для получения текущей экспериментальной версии нужно выполнить команду svn checkout http://svn.code.sf.net/p/cp2k/code/trun Настройка параметров компиляции производится с помощью файлов в подкаталоге cp2k/cp2k/arch полученного дерева исходников. Название файла в этом каталоге соответствует представляемой им "платформе" --- важнейшим параметрам системы: ОС, компилятор, набор инструкций процессора, а также способ параллелизации. CP2K поддерживает параллельность через MPI и через OpenMP, а также через комбинацию двух этих вариантов. По опыту работы на однопроцессорных многоядерных системах --- OpenMP в CP2K работает быстрее и [естественно] жрет меньше памяти, чем MPI. Потому править будем файл Linux-x86-64-gfortran.ssmp. Мой конфиг, удачно прошедший компиляцию, выглядит так: Без CUDA: С CUDA: Особо обращаю внимание, что $NVFLAGS обязательно должны содержать $(DFLAGS) --- инструкции по сборке этого не упоминают, но без этого компиляция не удастся. Поместив нужный файл в подкаталог cp2k/cp2k/arch, переходим в подкаталог cp2k/cp2k/makefiles и выполняем make ARCH=Linux-x86-64-gfortran VERSION=ssmp Если используете версию с CUDA, я бы рекомендовал отдельно собрать версию без CUDA, и держать рядом на всякий случай. Например, на карте с 1 Гб видеопамяти CP2K периодически ругается на невозможность выделить видеопамять и вылетает, приходится запускать без GPU. Названия версий стандартные, но принципиально не ограничены --- скажем, для конфигурации без CUDA можно создать конфигурационный файл Linux-x86-64-gfortran.ssmpnc (nc всмысле no-CUDA) или как-то так. Результат компиляции будет в подкаталоге cp2k/cp2k/exe, там будет ряд файлов, реально нужен только cp2k.ssmp (ну и cp2k.ssmpnc), который можно копировать куда угодно и использовать. Отдельное замечание об очистке каталога при неудачной сборке. Авторы наделали много разных опций для очистки, до конца там не совсем понятно, что следует использовать в каких случаях, я использую make clean ; make veryclean ; make realclean ; make distclean --- упорото, но вроде работает. |